Bacterial Endotoxins (LAL) Tests
The Bacterial Endotoxin Test also known as Limulus Amebocyte Lysate (LAL) Test, is an in vitro assay for the detection and quantitation of bacterial endotoxin in injectable drugs or solutions for parenteral administration, including biological products. The current USP General Chapter <85> “Bacterial Endotoxin Test” has been harmonized with the European and Japanese Pharmacopeia. Pacific BioLabs analysts are qualified to conduct the kinetic-chromogenic test. In this method the endotoxin content of the test material is determined by interpolation from a standard regression curve which is established each time the test is conducted.
The validation of the LAL test also known as Inhibition and Enhancement Test must be conducted for each drug formulation to determine the degree of product inhibition or enhancement of the assay. The validation must be conducted before the LAL test is used to assess the endotoxin content of any drug. All validation tests should be performed on undiluted product or on appropriate dilutions that do not exceed the Maximum Valid Dilution (MVD). The endotoxin limit of the drugs to be tested must be provided to Pacific BioLabs analysts by the sponsor at the time of sample submission in units such as EU/mL or EU/mg. The analysts need this limit to calculate the MVD for the validation test. Three production batches of each product must be tested for inhibition and enhancement.
A pyrogen is frequently described as a fever producing substance. The most potent pyrogens originate from gram negative bacteria, which are common water-borne organisms. Although not entirely accurate, the terms pyrogen and endotoxin are often used interchangeably. Detection of bacterial endotoxin contamination is essential to insure the safety of parenteral products. The Bacterial Endotoxins Test using Limulus Amebocyte Lysate (LAL) is recommended for the detection of endotoxins in pharmaceutical products. Human and animal injectable drugs (including biological products) must be tested for bacterial endotoxin.
The number of samples to be tested should be based on the manufacturing procedure and batch size. A minimum of three units, representing the beginning, middle, and end, of a production lot should be tested. These units can be tested individually or pooled.
The endotoxin limit for parenteral drugs, except those administered intrathecally, is defined as K/M, where K is equal to 5.0EU/Kg and M is the maximum dose per patient weight in Kg. For parenteral drugs that have an intrathecal route of administration, K is equal to 0.2 EU/Kg.
FDA has published guidelines outlining validation procedures for endotoxin testing of finished products using the LAL test. This document is titled Guideline on the Validation of the Limulus Amebocyte Lysate Test for Human and Animal Parenteral Drugs, Biological Products and Medical Devices and can be downloaded at www.fda.gov
Because water is a source of pyrogens, it is important to routinely monitor water systems using the bacterial endotoxins test. For process water samples, the amount of sample required is 10 mL. Please contact Pacific BioLabs if you need sample collection containers.
Kinetic Chromogenic LAL testing available at PBL:
- LAL Validation – Inhibition and Enhancement
- Pharmaceutical Powders
- Pharmaceutical Liquids
- Water
Microbial Limits Test (MLT)
Microbial Limits Testing (MLT) is used to determine whether a product complies with an established specification for microbial quality. This test is designed to estimate the number of viable aerobic microorganisms present in a product, and defines designated microbial species for products from the raw materials stage to a final manufactured product.
The MLT is performed under controlled conditions to avoid accidental contamination of the product being tested, and is subdivided into 2 tests as shown below:
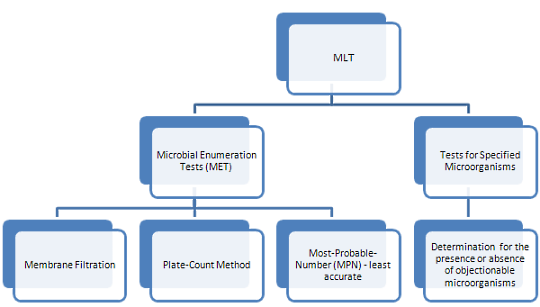
Microbial Enumeration Test (MET)
The MET is a quantitative enumeration of mesophilic bacteria and fungi that grow under aerobic conditions. Results can be interpreted as follows:
- Total Aerobic Microbial Count (TAMC) – number of colony forming units (cfu) found using Tryptic Soy Agar (TSA)
- Total Combined Yeasts and Molds Count (TYMC) – number of cfu found using Sabouraud Dextrose Agar (SDA)
The acceptance criteria for microbiological quality of a product is prescribed and interpreted as follows:
- 10^1 cfu: maximum acceptable count = 20
- 10^2 cfu: maximum acceptable count = 200
- 10^3 cfu: maximum acceptable count = 2000
There are 3 methods used in enumerating the microbial content of a product. The choice of a method should be based on the nature of the product and the required limit of microorganisms.
- Membrane Filtration uses filtration apparatus.
- Total Plate-Count Method (TPC) involves direct plating.
- Most-Probable-Number (MPN) Method is the least accurate method for microbial counts, however, it can be the most appropriate method for certain products with very low bioburden.
Tests for Specified Microorganisms
This is a screening test to detect the absence or presence of specified microorganisms in non-sterile products. These organism groups are:
- Bile-Tolerant Gram Negative Bacteria
- Escherichia coli and Salmonella
- Pseudomonas aeruginosa and Staphylococcus aureus
- Clostridia and Candida albicans
Testing Considerations
Prior to performing testing, the following procedures should be followed:
- Growth Promotion
- Suitability of the Counting Method (Validation)
- Suitability Tests for Specified Microorganisms (Validation)
These test results can only be shown to be valid if it can be demonstrated that the test article does not inhibit microbial growth. Any product with an antimicrobial component needs to be neutralized to remove its antimicrobial activity.
Additionally, it must be shown that the test to be used can actually detect microorganisms in the presence of the product to be tested. Suitability must be confirmed if a change in testing performance or a change in the product is introduced that may affect the outcome of the test.
Acceptance criteria for the Microbiological Quality of non-sterile products depend on the nature and route of administration of the product in its final form. Manufacturers should ensure a low bioburden of the final product by implementing Good Manufacturing Practice during the manufacture, storage and distribution of their products.
Harmonization of Standards
International Harmonization of Standards provides information on the harmonization of various general chapters from Japanese, United States, European and British pharmacopoeias. The purpose of harmonization is to arrive at interchangeable methods or requirements so that a demonstration of compliance using a general chapter from any one of the 3 pharmacopoeias will give the same result. Harmonization also aims to arrive at identical requirements for all attributes of a specified product.
The table below shows the different chapters from each standard that were harmonized
Tests | Japanese Pharmacopoeia (JP) | US Pharmacopoeia (USP) | European Pharmacopoeia (EP) | British Pharmacopoeia (BP) |
Examination of Non-Sterile products: Microbial Enumeration Test (MET) | Japanese Pharmacopoeia XV 1st Supplement | USP<61> | EP chapter 2.6.12 | BP chapter B2 |
Examination of Non-Sterile Products: Tests for Specified Microorganisms | Japanese Pharmacopoeia XV 1st supplement | USP<62> | EP chapter 2.6.13 | BP chapter B1 |
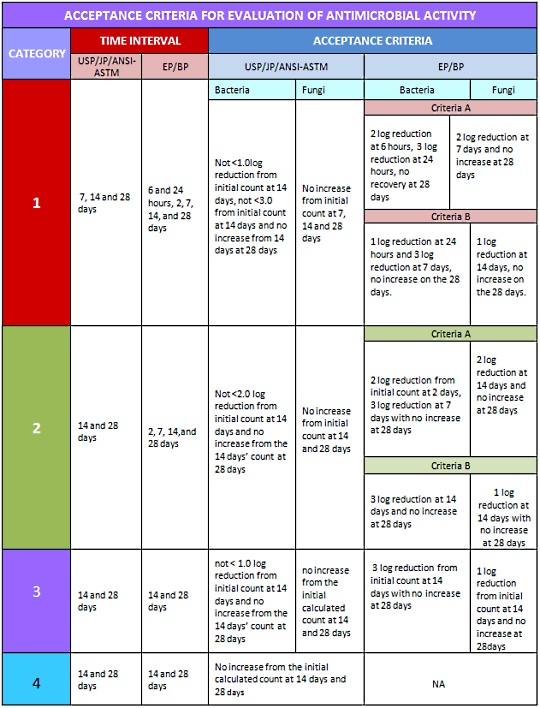
Antimicrobial Effectiveness Test (AET) – USP
Antimicrobial preservatives are often added to compendial products for the following reasons:
- To prevent proliferation or limit microbial contamination that may occur subsequent to the manufacturing process.
- To inhibit growth of microorganisms during normal conditions of storage and use when microorganisms might be introduced inadvertently from repeated withdrawing of individual doses from containment.
- To prolong the life span of the product.
*Please bear in mind that although preservatives can provide protection from microbial contamination or proliferation, antimicrobial preservatives must not be used as a substitute for Good Manufacturing Practice.
Antimicrobial Effectiveness Testing is performed to support a claim that the added preservative in an item provides adequate protection from adverse effects that may arise from microbial contamination or proliferation during storage and use. This test is not intended to be used for routine control purposes but it instead designed to put a challenge on a compendial preparation in its final container with a prescribed inoculums of suitable microorganisms.
The AET is also known as Efficacy of Antimicrobial Preservation (EAP) in the European and British Pharmacopeia, and as the Preservatives-Effectiveness Test(PET) in the Japanese Pharmacopeia.
The USP and other compendia separate articles to be tested for antimicrobial effectiveness into 4 different categories based on their route of administration. Each category has its own acceptable criteria for tested microorganisms as shown in the table below: